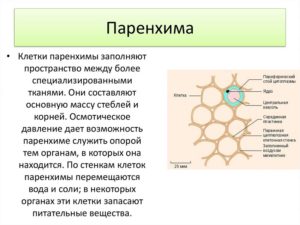
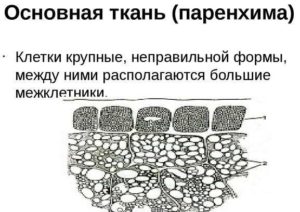
Паренхиматозные клетки это
Содержание
Паренхиматозные белковые дистрофии
Сохрани ссылку в одной из сетей:
ПАРЕНХИМАТОЗНЫЕДИСТРОФИИ (ПД)
Определение: ПД– это дистрофии, которые развиваютсяв клетках паренхиматозных органов(нефроциты, гепатоциты, кардиомиоциты).
Классификация. Повиду нарушенного обменасреди ПД выделяют:
- белковые,
- жировые,
- углеводные.
Паренхиматозные белковыедистрофии
Среди этих видов дистрофийвыделяют:
- гиалиново-капельную,
- гидропическую,
- роговую.
Гиалиново-капельнаядистрофия
Определение: этотяжелая необратимая белковаяпаренхиматозная дистрофия, при которойв цитоплазме появляются крупные каплипохожие на гиалин.
Локализация:нефроциты почек, гепатоциты печени,кардиомиоциты миокарда
Патоморфология:макро – изменений нет, микро – внефроцитах находят крупные гиалиноподобныекапли и разрушенные ультраструктурыклетки, в гепатоцитах находят аномальныйбелок (алкогольный гиалин) или тельцаМэллори.
Этиология: впочках – это болезни почек (гломерулонефриты),в печени – алкогольный гепатит.
Патогенез: впочках – это инфильтрация, в печени –аномальный синтез.
Исход: неблагоприятный– сухой некроз клетки.
Функция:в почках это проявляется белком в моче(протеинурия),цилиндрами в моче (цилиндрурия), потерябелков плазмы крови (гипопротеинемия),нарушения ее электролитного баланса,в печени нарушением многих функций.
Гидропическаядистрофия (баллонная дистрофия)
Определение: этотяжелая необратимая белковаяпаренхиматозная дистрофия, характеризующаясяпоявлением в клетке вакуолей, наполненныхжидкостью.
Локализация: эпителийкожи, нефроциты почки, гепатоциты печени,кардиомиоциты сердца, миоциты, нервныеклетки, клетки коры надпочечников.
Патоморфология:макро – изменений нет, микро – клеткиувеличены, в цитоплазме вакуоли сжидкостью. Ядро – на периферии. Прирезко выраженной дистрофии клеткипохожи на баллон – это баллонноядистрофия.
Этиология: в почках– это болезни почек (гломерулонефриты,амилоидоз почек, сахарный диабет), впечени – вирусные и токсические гепатиты,в коже – оспа, отеки различного генеза,в мышцах – сепсис, в сердце – болезнисердца (ишемия).
Патогенез: нарушениепроницаемости мембран клетки иинфильтрация жидкости в цитоплазму.
Исход: неблагоприятный– влажный некроз.
Функция: резкоеснижение функции органов, может приводитьк недостаточности органов.
Роговаядистрофия (патологическое ороговение)
Определение: этадистрофия характеризуется образованиембольшого количества рогового веществав ороговевающем эпителии (кожа) –гиперкератоз, ихтиоз, илипроисходит ороговение эпителия слизистыхоболочек – лейкоплакия,образование «раковых жемчужин» вплоскоклеточном раке.
Этиология: нарушениеразвития кожи, хроническое воспаление,вирусные инфекции, авитаминозы.
Исход: при устранениипричины в начале развития дистрофииприводит к восстановлению ткани, впротивном случае гибель клеток.
Значение:определяется ее степенью, распространенностьюи длительностью. Приводит к раковойопухоли, врожденный ихтиоз несовместимс жизнью.
Паренхиматозныежировые дистрофии
Определение: этодистрофии, при которых в цитоплазмеклеток (нефроциты, гепатоциты,кардиомиоциты) происходит повышенноенакопление липидов.
Локализация: миокард,печень, почки.
Сердце:макро – орган увеличен,консистенция миокарда дряблая, камерыего расширены, цвет на разрезеглинисто-желтый, со стороны эндокардана трабекулярных мышцах заметны полоски– “тигровое сердце”.Микро – в цитоплазме кардиомиоцитовпоявляются капли жира небольших размеров(пылевидное ожирение),или более крупные (мелкокапельноеожирение). Окраска – судан3.
Патогенез: декомпозицияи инфильтрация
Исход:на ранних стадиях – обратимый процесс,при выраженных изменениях ведет кнекрозу клетки.
Функция:жировая дистрофия миокарда проявляетсяу больных хронической сердечно-сосудистойнедостаточностью.
Печень:макро – орган увеличен, консистенция– дряблая, цвет – желто-коричневый, видна разрезе – “гусинаяпечень”. Микро – в клеткахсначала мелкие капли жира (мелкокапельноеожирение), которые потомсливаются в крупные капли (крупнокапельноеожирение). Ядро смещаетсяна периферию и клетка имеет округлуюформу. По своему виду напоминает жировыеклетки. Окраска – судан 3.
Этилогия: интоксикации(алкоголь, лекарства, промышленные яды),нарушения питания (недостаток белка ивитаминов в пище), сахарный диабет, общееожирение.
Патогенез:инфильтрация и трансформация.
Исход: наранних стадиях процесс может бытьобратим, на поздних стадиях ведет кразвитию цирроза печени.
Функция: нарушениефункции печени.
Почки:макро – орган увеличен, дряблые (при сочетании с амилоидозом плотные),корковое вещество набухшее, серое сжелтым крапом, заметным на поверхностии разрезе.
Патогенез: инфильтрация.
Источник: https://gigabaza.ru/doc/92390.html
Паренхиматозные дистрофии. Патологическая анатомия
Иногда в клинической практике встречается такое явление, как паренхиматозные дистрофии. Патологическая анатомия относит их к нарушениям обмена в клетках.
Если говорить простым языком, то в органе нарушается процесс питания и накопления полезных веществ, что приводит к морфологическим (визуальным) изменениям. Выявить такую патологию можно на секции или после серии высокоспецифических тестов.
Паренхиматозные и стромально-сосудистые дистрофии лежат в основе многих летальных заболеваний.
Определение
Паренхиматозные дистрофии – это патологические процессы, которые ведут к изменениям структуры клеток органов. Среди механизмов развития заболевания выделяют расстройства саморегуляции клетки с энергетическим дефицитом, ферментопатии, дисциркуляторные расстройства (кровь, лимфа, интерстиций, межклеточная жидкость), эндокринные и церебральные дистрофии.
Различают несколько механизмов дистрофии:
– инфильтрацию, то есть избыточный транспорт продуктов обмена из крови внутрь клетки или межклеточное пространство, обусловленный сбоем в ферментных системах организма;
– декомпозиция, или фанероз, представляет собой распад внутриклеточных структур, который приводит к нарушению метаболизма и накоплению недоокисленных продуктов обмена веществ;
– извращенный синтез веществ, которые в норме клетка не воспроизводит;
– трансформация поступающих в клетку питательных веществ для построения какого-то одного вида конечных продуктов (белков, жиров или углеводов).
Классификация
Патоморфологи выделяют следующие виды паренхиматозных дистрофий:
1. В зависимости от морфологических изменений:
– чисто паренхиматозные;
– стромально-сосудистые;
– смешанные.
2. По виду накапливаемых веществ:
– белковые или диспротеинозы;
– жировые или липидозы;
– углеводные;
– минеральные.
3. По распространенности процесса:
– системные;
– местные.
4. По времени появления:
– приобретенные;
– врожденные.
Те или иные паренхиматозные дистрофии патологическая анатомия определяет не только по повреждающему агенту, но и по специфике пораженных клеток.
Переход одной дистрофии в другую теоретически возможен, но практически возможна только сочетанная патология.
Паренхиматозные дистрофии – это суть процесса, происходящего в клетке, но только часть клинического синдрома, который охватывает морфологическую и функциональную недостаточность определенного органа.
Диспротеинозы
Человеческое тело по большей части состоит из белков и воды. Белковые молекулы являются составляющей клеточных стенок, мембраны митохондрий и других органелл, кроме того, они находятся в свободном состоянии в цитоплазме. Как правило, это ферменты.
Диспротеинозом иначе называют такую патологию, как паренхиматозная белковая дистрофия. И его суть состоит в том, что клеточные белки меняют свои свойства, а так же подвергаются структурным изменениям, таким как денатурация или колликвация.
К белковым паренхиматозным дистрофиям относят гиалиново-капельную, гидропическую, роговую и зернистую дистрофии.
О первых трех будет написано подробнее, а вот последняя, зернистая, характеризуется тем, что в клетках накапливаются зерна белка, из-за чего клетки растягиваются, а орган увеличивается, становится рыхлым, тусклым. Именно поэтому зернистую дистрофию еще называют тусклым набуханием.Но у ученых есть сомнения, что это паренхиматозная дистрофия. Патанатомия данного процесса такова, что за зерна можно принять компенсаторно увеличенные клеточные структуры, как ответ на функциональное напряжение.
При этом виде дистрофий в клетках появляются большие гиалиновые капли, которые со временем сливаются между собой и заполняют все внутреннее пространство клетки, вытесняя органеллы или разрушая их. Это приводит к потере функции и даже гибели клетки. Чаще всего заболевание встречается в почечной ткани, реже в печени и сердце.
Во время цитологического исследования после биопсии почек, помимо накопления гиалина в нефроцитах, обнаруживают деструкцию всех клеточных элементов.
Это явление появляется, если у пациента развивается вакуолярно-лизосомальная недостаточность, которая приводит к уменьшению реабсорбции белков из первичной мочи. Чаще всего данная патология встречается при нефротическом синдроме.
Наиболее часты диагнозы таких пациентов – гломерулонефрит и амилоидоз почек. Внешний вид органа при гиалиново-капельной дистрофии не изменяется.
В клетках печение дело обстоит несколько иначе. Во время микроскопии в них находят тельца Мэллори, состоящие из фибрилл и алкогольного гиалина. Их появление связано с болезнью Вильсона-Коновалова, алкогольным гепатитом, а также с билиарным и индийским циррозом. Исход этого процесса неблагоприятный – некроз клеток печени, утрата ее функции.
Гидропическая дистрофия
Этот вид дистрофий отличается от остальных тем, что в пораженных клетках появляются новые органеллы, наполненные жидкостью. Чаще всего такое явление можно заметить в коже и канальцах почек, в клетках печени, мышц и надпочечников.
Микроскопически клетки увеличены, их цитоплазму заполняют вакуоли с прозрачным жидким содержимым. Ядро смещается или лизируется, остальные структуры элиминируются. В конечном итоге клетка представляет собой «баллон», заполненный водой. Поэтому гидропическую дистрофию иногда называют баллонной.
Макроскопически органы практически не изменяются. Механизм развития этого заболевания – нарушение коллоидно-осмотического давления в клетке и в межклеточном пространстве.
Из-за этого проницаемость клеток увеличивается, мембраны их распадаются и клетки погибают. Причинами таких химических изменений может стать гломерулонефрит, сахарный диабет, амилоидоз почек.
В печени изменению клеток способствуют вирусные и токсические гепатиты. На коже гидропическая дистрофия может быть вызвана вирусом натуральной оспы.Заканчивается этот патологический процесс фокальным или тотальным некрозом, поэтому морфология и функция органов быстро ухудшается.
Роговая дистрофия
Патологическое ороговение органов – это чрезмерное накопление рогового вещества в поверхностных слоях кожи, например, гиперкератоз или ихтиоз, а также появление рогового вещества там, где, как правило, его быть не должно – на слизистых оболочках (лейкоплакия, плоскоклеточный рак). Это процесс может быть как локальным, так и тотальным.
Причинами этого типа заболеваний могут быть нарушения эктодермального зачатка в процессе эмбриогенеза, хронические воспалительные изменения тканей, вирусные инфекции и недостаток витаминов.
Если лечение начать сразу после появления первых симптомов, то ткани еще могут восстановиться, но в далеко зашедших случаях выздоровление уже невозможно. Длительно существующие участки роговой дистрофии могут переродиться в рак кожи, а врожденный ихтиоз несовместим с жизнью плода.
Наследственные дистрофии
Наследственные паренхиматозные дистрофии возникают из-за врожденных ферментопатий. Эти заболевания иначе называют болезни накопления, так как из-за нарушения метаболизма, продукты обмена веществ накапливаются в клетках и жидкостях организма, отравляя его. Наиболее известными представителями этой группы являются фенилкетонурия, тирозиноз, и цистиноз.
Органами-мишенями для фенилкетонурии являются центральная нервная система, мышцы, кожа и жидкости (кровь, моча). Продукты обмена при тирозинозе накапливаются в клетках печени, почек и костях. Цистиноз также поражает печень и почки, но, кроме них, страдает селезенка, глазные яблоки, костный мозг, лимфатическая система и кожа.
Липиды содержатся в каждой клетке, они могут находиться как отдельно, так и в комплексе с белками и быть структурными единицами мембраны клеток, а также других ультраструктур. Кроме того, в цитоплазме находится глицерин и жирные кислоты.
Для того чтобы обнаружить их в тканях используются специальные методы фиксирования и окрашивания, например суданом черным или красным, осмиевой кислотой, сульфатом нильского голубого.
После специфической подготовки препараты тщательно осматривают в электронный микроскоп.
Паренхиматозная жировая дистрофия проявляется в виде чрезмерного накопления жиров там, где они должны быть, и появления липидов там, где их быть не должно. Как правило, накапливаются нейтральные жиры. Органы мишени те же, что и при белковой дистрофии – сердце, почки и печень.
Жирова паренхиматозная дистрофия миокарда начинается с появления в миоцитах очень мелких капелек жира, т. н. пылевидное ожирение.
Если процесс не останавливается на этом этапе, то со временем капли сливаются и становятся больше, пока не займут всю цитоплазму. Органеллы при этом распадаются, исчерченность мышечных волокон пропадает.Заболевание локально проявляется около венозного сосудистого русла.
Макроскопически паренхиматозная жировая дистрофия проявляется по-разному, все зависит от стадии процесса. В самом начале диагноз можно поставить только под микроскопом, но со временем сердце увеличивается за счет растягивания камер, стенки его становятся тонкими и дряблыми, при разрезе миокарда видны грязно-желтые полосы. Патофизиологи придумали название этому органу: «тигровое сердце».
Жировая дистрофия паренхиматозных органов развивается по трем основным механизмам.
- Повышенное поступление свободных жирных кислот в клетки миокарда.
- Нарушение жирового обмена.
- Распад липопротеидных структур внутри клетки.
Чаще всего эти механизмы запускаются во время гипоксии, инфекции (дифтерия, туберкулез, сепсис) и интоксикации организма хлором, фосфором или мышьяком.
Как правило, жировая дистрофия обратима, а нарушения клеточных структур восстанавливаются со временем. Но если процесс сильно запущен, то все заканчивается гибелью ткани и органа. Клиницисты различают следующие заболевания, связанные с накоплением жиров в клетках:
– болезнь Гоше;
– болезнь Тея-Сакса;
– болезнь Ниманна-Пика и другие.
Углеводные дистрофии
Все углеводы, которые находятся в организме, можно разделить на полисахариды (самым распространенным из которых является гликоген), гликозаминогликаны (мукополисахариды: гиалуроновая и хондроитинсерная кислоты, гепарин) и гликопротеиды (муцины, т.е. слизь, и мукоиды).
Для того чтобы выявить углеводы в клетках организма, проводят специфический тест – ШИК-реакцию. Суть ее в том, что ткань обрабатывают йодной кислотой, а потом фуксином. И все альдегиды становятся красными.
Если нужно выделить гликоген, то к реактивам добавляют амилазу. Гликозаминогликаны и гликопротеиды окрашиваются метиленовым синим.
Паренхиматозные углеводные дистрофии связаны, как правило, с нарушением обмена гликогена и гликопротеидов.
Нарушение обмена гликогена
Гликоген – это запасы организма на «черный голодный день». Основную их часть он хранит в печени и мышцах и расходует эту энергию очень экономно. Регулирование обмена углеводов происходит через нейроэндокринную систему. роль отведена, как водится, гипоталамо-гипофизарной системе. В ней вырабатываются тропные гормоны, которые контролируют все остальные железы внутренней секреции.
Нарушением гликогенового обмена является увеличение или снижение его количества в тканях, а также появление там, где он быть не должен. Наиболее ярко такие изменения проявляются при сахарном диабете либо наследственных гликогенозах.
Патогенез сахарного диабета довольно хорошо изучен: клетки поджелудочной железы перестают вырабатывать инсулин в необходимом количестве, и энергетические запасы клеток быстро истощаются, так как глюкоза не накапливается в тканях, а выводится из организма с мочой.
Организм «вскрывает» свои резервы, и в первую очередь развивается паренхиматозная дистрофия печени. В ядрах гепатоцитов появляются светлые промежутки, и они становятся светлыми. Поэтому их еще называют «пустые ядра».Наследственные гликогенозы вызваны нехваткой или отсутствием ферментов, участвующих в накоплении гликогена. В настоящее время известны 6 таких болезней:
– болезнь Гирке;
– болезнь Помпе;
– болезнь Мак-Ардля;
– болезнь Герса;
– болезнь Форбса-Кори;
– болезнь Андерсена.
Их дифференциальная диагностика возможна после биопсии печени и использования гистоферментного анализа.
Нарушение обмена гликопротеинов
Это паренхиматозные дистрофии, вызванные накоплением в тканях муцинов или мукоидов. Иначе эти дистрофии еще называют слизистыми или слизеподобными, из-за характерной консистенции включений. Иногда накапливаются на истинные муцины, а только похожие на них вещества, которые могут уплотняться. В таком случае идет речь о коллоидной дистрофии.
Микроскопия ткани позволяет определить не только факт наличия слизи, но и ее свойства. Из-за того, что остатки клеток, а также вязкий секрет препятствует нормальному оттоку жидкости из желез, образуются кисты, а содержимое их имеет тенденцию к воспалению.
Причины этого вида дистрофий могут быть самые разные, но чаще всего это катаральное воспаление слизистых. Кроме того, если наследственное заболевание, патогенетическая картина которого хорошо вписывается в определение слизистая дистрофия. Это муковисцидоз. Поражается поджелудочная железа, кишечная трубка, мочевыводящий тракт, желчные протоки, потовые и слюнные железы.
Разрешение данного вида заболеваний зависит от количества слизи и длительности ее выделения. Чем меньше времени прошло от начала патологического процесса, тем более вероятно, что слизистая восстановится полностью. Но в некоторых случаях наблюдается слущивание эпителия, склероз и нарушение функции пораженного органа.
Источник: https://FB.ru/article/274311/parenhimatoznyie-distrofii-patologicheskaya-anatomiya
Дистрофия. Виды и классификация паренхиматозных дистрофий

Подробности
Дистрофия – сложный патологический процесс, в основе которого лежит нарушение тканевого метаболизма, ведущее к структурным изменениям.
Трофика – совокупность механизмов , определяющих метаболизм и структурную организацию клетки (ткани), необходимых для выполнения специализированной функции.
Причины дистрофий:
1) расстройства ауторегуляции клетки, которые могут быть вызваны гиперфункцией, токсическими веществами, радиацией, недостаточностью фермента и т.д.
2) нарушение функции транспортных систем, обеспечивающих метаболизм и структурную сохранность тканей, вызывают гипоксию.
3) нарушение эндокринной, нервной регуляции
1) инфильтрация
Избыточное накопление вещества (нормального, не аномального) в результате избыточного синтеза.
Пример: жировой гепатоз печени, гемосидероз почки.
2) декомпозиция (фанероз)
Распад ультраструктур клеток и межклеточного вещества, ведущий к нарушению тканевого метаболизма и накоплению продуктов нарушенного обмена в ткани.
3) извращенный синтез
Синтез аномальных продуктов. К ним относятся: синтез аномального белка амилоида в клетке, синтез белка алкогольного гиалина гепатоцитом.
4) трансформация
Образование продуктов одного вида обмена из общих исходных продуктов, которые идут на построение БЖУ.
Классификации дистрофии
В классификации придерживаются нескольких принципов. Выделяют дистрофии:
1) по преобладанию морфологических изменений в тканевых структурах: паренхиматозные, смешанные, мезенхимальные (стромально-сосудистые)
2) по преобладанию нарушений того или иного вида обмена: белковые, жировые, углеводные, минеральные.
3) в зависимости от влияния генетических факторов: приобретенные, наследственные.
4) по локализации: местные, общие.
Паренхиматозные дистрофии.
Проявления нарушений обмена в высокоспециализированных в функциональном отношении клетках.
1) Паренхиматозные белковые дистрофии (диспротеинозы)
Сущность таких дистрофий состоит в изменении физико-химических и морфологических свойства белков клетки: они подвергаются денатурации и коагуляции или колликвации, что ведет к гидратации цитоплазмы. В тех случаях, когда нарушатся связи белков с липидами, возникает деструкция мембранных структур клетки.
Нарушение обмена белков часто сочетается с расстройствами работы Na-K-помпы: что ведет к накоплению ионов Nа и набуханию клетки. Такой патологический процесс называется гидропической дистрофией.
Виды:
-зернистая
Обратима, выглядит как накопление мелких зерен белка в цитоплазме. Органы увеличиваются в размерах, становятся дряблыми и тусклыми.
-гиалиново-капельная
В цитоплазме появляются крупные гиалиноподобные белковые капли, сливающиеся между собой и заполняющие тело клетки. В ряде случаев завершается фокальным коагуляционным некрозом клетки.
Часто встречается в почках, редко – в печени и миокарде.
В почках при исследовании накопление капель находят в нефроцитах. Накопление часто отмечается при нефротическом синдроме, так как в основе этой дистрофии лежит недостаточность вакуолярно-лизосомального аппарата эпителия проксимального канальца, в котором в норме реабсорбируются белки. Именно поэтому в моче появляется белок (протеинурия) и цилиндры (цилиндрурия).
Внешний вид не имеет каких-либо характерных черт.
В печени при микроскопии обнаруживаются тельца Мэлори, состоящие из фибрилл и алкогольного гиалина. Появление таких капель – проявление извращенной синтетической функции гепатоцита, что встречается при алкогольном гепатите, первичном билиарном циррозе. Внешний вид печени различен.
Исход гиалиново-капельной дистрофии неблагоприятен, она ведет к некрозу клетки.
-гидропическая дистрофия
Характеризуется появление в клетке вакуолей, наполненных цитоплазматической жидкостью. Наблюдается чаще в эпителии кожи и почечных канальцев, в гепатоцитах и миоцитах.
Паренхиматозные клетки увеличены в объеме, их цитоплазма заполнена вакуолями, содержащими прозрачную жидкость. Затем клетка превращается в огромный баллон (вся клетка стала большой вакуолью) – фокальный колликвационный некроз. Внешний вид тканей мало изменяется.
Большую роль в механизме развития играет нарушение проницаемости мембраны, которое ведет к закислению цитоплазмы, активации гидролитических ферментов лизосом, которые разрывают внутримолекулярные связи с присоединением воды.
Причины: в почках – повреждение почечного фильтра, что ведет к гиперфильтрафии, в печени – гепатиты различной этиологии, в эпидермисе – отек, инфекция.Исход такой дистрофии, как правило, неблагоприятный – она завершается фокальным коагуляционным некрозом.
-роговая дистрофия
Характеризуется избыточным образованием рогового вещества в ороговевающем эпителии (гиперкератоз, ихтиоз) или образование рогового вещества там, где в норме его не бывает (патологическое ороговение на слизистых оболочках). Причины разнообразны: нарушения развития кожи, хроническое воспаление, авитаминозы и т.д.
Исход: иногда при устранении причины происходит восстановление ткани, однако в запущенных случаях наступает гибель клеток.
– наследственные нарушения обмена аминокислот
Так называемые болезни накопления, в основе которых лежит нарушение внутриклеточного метаболизма ряда аминокислот в результате наследственной недостаточности метаболизирующих ферментов.
А) цистиноз. Науке еще не известно, недостаточность какого фермента приводит к этому заболеванию. АК накапливается в печени, почках, селезенке, глазах, костном мозге, коже.
Б) тирозиноз. Возникает при дефиците тирозинаминотрансферазы. Накапливается в печени, почках, костях.
В) фенилпировиноградная олигофрения. Возникает при дефиците фенилаланин-4-гидроксилазы и накапливается в нервной системе, мышцах и крови.
2) Паренхиматозные жировые дистрофии (липидозы)
Нарушения обмена цитоплазматических липидов могут проявляться в увеличении их содержания в клетках, где они обнаруживаются и в норме., в появлении липидов там, где они обычно не встречаются, и в образовании жиров необычного химического состава.
–нарушения обмена липидов
В печени жировая дистрофия проявляется резким увеличением содержания жиров в гепатоцитах и изменением их состава.
В клетках печени сначала появляются гранулы липидов (пылевидное ожирение), затем мелкие капли (мелкокапельное ожирение), которые затем сливаются в крупные капли (крупнокапельное) или в одну жировую вакуоль. Печень увеличена, дряблая и охряно-желтого цвета.
Среди механизмов жировой дистрофии печени различают, чрезмерное поступление в гепатоциты жирных кислот или повышенный их синтез этими клетками, воздействие токсических веществ, блокирующих окисление жирных кислот и синтез липопротеидов в гепатоцитах, недостаточное поступление в печеночные клетки аминокислот, необходимых для синтеза. Итак, ЖДП возникает в результате: липопротеидемии (алкоголизм, сахарный диабет, общее ожирение), гепатотропных интоксикациях (этанол, хлороформ), нарушения питания.
Жировая дистрофия миокарда возникает вследствие гипоксии и интоксикации. Механизм развития связан со снижением окисления жирных кислот из-за деструкции митохондрий под действием гипоксии или токсина.
При макроскопическом исследовании размеры сердца увеличены, сердечная мышца глиняно-желтого цвета. Миокард похож на шкуру тигра – бело-желтая исчерченность. Липиды определяются в виде мелких капель.
Причины жировой дистрофии разнообразны. Они могут быть связаны с кислородным голоданием (потому часто встречается при заболеваниях ССС), инфекциями и интоксикациями, авитаминозами и односторонним питанием.Исход жировой дистрофии зависит от ее степени. Если не сопровождается грубым поломом клеточных структур, то она обратима.
-наследственные ферментопатии
Возникают вследствие наследственного дефицита ферментов, участвующих в метаболизме липидов.
А) болезнь Гоше при дефиците глюкоцереброзидазы. Липид накапливается в печени, селезенке, костном мозге.
Б) болезнь Ниманна–Пика при дефиците сфингомиелиназы. Накопление в печени, селезенке, костном мозге.
В) болезнь Сакса при дефиците кислой галактозидазы.
Г) болезнь Нормана–Ландинга при дефиците бета-галактозидазы.
-углеводные дистрофии, связанные с нарушением обмена гликогена
Нарушения содержания гликогена проявляется в уменьшении или увеличении количества его в тканях и появлении там, где он обычно не выявляется.
При сахарном диабете происходит недостаточное использование глюкозы тканями, увеличение ее содержания в крови и выведение с мочой. Тканевые запасы гликогена резко уменьшаются. В печени нарушается синтез гликогена, что ведет к ее инфильтрации жирами и жировой дистрофии печени.
В почках при сахарном диабете происходят следующие изменения: гликогенная инфильтрация эпителия канальцев.
-наследственные гликогенозы
а) 1 типа – болезнь Гирке – дефицит глюкозо-6-фосфатазы
б) 2 типа – болезнь Помпе – дефицит кислой альфа-1,4-глюкозидазы
в) 3 типа – болезнь Форбса – дефицит амило-1,6-глюкозидазы
г) 4 типа – болезнь Андресона – дефицит амило-(1,4-1,6)-трансглюкозидазы
д) 5 типа – болезнь Мак-Ардля – дефицит миофосфорилазы
е) 6 типа – болезнь Герса – дефицит фосфорилазы печени
При болезнях 1,2,5,6 типов структура гликогена не нарушена.
–углеводные дистрофии, связанные с нарушениями обмена гликопротеидов
В клетках или межклеточном веществе происходит накопление муцинов и мукоидов, называемых также слизистыми или слизеподобными вещества.
Многие секретирующие клетки погибают и десквамируются, выводные протоки желез обтурируются слизью, что ведет к развитию кист.
Причины разнообразны, но чаще всего – воспаление слизистых оболочек в результате действия различных патогенных раздражителей.
Источник: http://fundamed.ru/patan/152-distrofiya-vidy-i-klassifikatsiya-parenkhimatoznykh-distrofij.html
Жировые дистрофии (липидозы)

Жировые дистрофии связаны с избыточным накоплением в цитоплазме паренхиматозных клеток липидов (нейтральных жиров, триглицеридов, фосфолипидов, холестерина), либо с появлением их в тех клетках, где они в норме не встречаются, либо с появлением в цитоплазме клеток липидов аномального состава. Потребность человека в жирах составляет 80—100 г в сутки.
Основные функции липидовв организме:
- структурная — липиды составляют основу клеточных мембран;
- регулирующая;
- энергообеспечивающая, поскольку липиды являются одним из главных источников энергии.
В зависимости от клинических проявлений выделяют:
- липидозы;
- ожирение;
- истощение.
Причинами приобретенных липидозов наиболее часто служат гипоксия и различные интоксикации. Поэтому жировые дистрофии являются компонентом заболеваний, сопровождающихся ки-i слородным голоданием.
— ишемической болезни сердца, гипертонической болезни, пороков сердца, хронических заболеваний легких (бронхоэктатическая болезнь, туберкулез, эмфизема легких). приводящих к развитию легочно-сердечной недостаточности.
Кроме того, жировую дистрофию вызывают различные инфекции и интоксикации, которые сопровождаются как гипоксией, так и блокадой токсинами ферментов, катализирующих в клетках метаболизм липидов. Липидозы иногда могут быть связаны с недостатком витаминов и некоторых аминокислот.
В патологии наибольшее значение имеют жировые дистрофии миокарда, печени и почек.
Рис. 3. Жировая дистрофия миокарда. а — кардиомиоциты с жировым включениями; б — венулы; в — кардиомиоциты, свободные от жировых включений.
Жировая дистрофия миокарда развивается путем декомпозиции жиробелковых комплексов мембран внутриклеточных структур, а также в результате инфильтрации кардиомиоцитов липидами.
Вне зависимости от механизма дистрофии в клетках миокарда вначале появляются мелкие включения жира (пылевидное ожирение), затем они сливаются в капли (мелкокапельное ожирение), которые постепенно заполняют всю саркоплазму и могут приводить к гибели клеток (рис. 3).
на для резкого снижения функции сердца и развития сердечной недостаточности.
Жировая дистрофия печени, или жировой гепатоз
Рис. 4. Жировая дистрофия миокарда («тигровое сердце»). Под эндокардом видны желто-белые полоски, соответствующие участкам включения липидов в кардиомиоцитах.
Вместе с тем при многих интоксикациях и инфекциях возможен механизм декомпозиции мембран внутриклеточных структур с распадом их жиробелковых комплексов. Наконец, жировой гепатоз развивается в результате трансформации белков и углеводов в липиды, что наблюдается, например, при хронической алкогольной интоксикации.
В любом случае в цитоплазме гепатоцитов, в основном периферии печеночных долек, развивается вначале пылевидное ожирение, которое трансформируется в мелкокапельное, а затем — в крупнокапельное. При этом ядро и внутриклеточные структуры оттесняются на периферию клеток, которые нередко гибнут. В этих случаях жировые включения погибших гепатоцитов сливаются, образуя жировые кисты.
Макроскопические изменения печени зависят от степени выраженности дистрофии. В тяжелых случаях, например при алкоголизме, печень увеличена в размере, дряблая, на разрезе охряного цвета — «гусиная печень». При менее выраженной жировой дистрофии печень также увеличена в размере, на разрезе желтовато-серого цвета.При жировых гепатозах функция печени долго сохраняется, однако по мере прогрессирования основного заболевания и жировой дистрофии она снижается, иногда весьма значительно.
Жировая дистрофия почек развивается путем инфильтрации эпителия канальцев при гиперлипидемии, наблюдающейся, в частности, при нефротическом синдроме.
В этой ситуации липиды оказываются в первичной моче (гиперлипидурия) и усиленно реабсорбируются клетками эпителия канальцев, но в таких больших количествах, что эти клетки не способны метаболизировать попавшие в них липиды — развивается мелкокапельное ожирение эпителия канальцев. Обычно оно сочетается с их гиалиново-капельной дистрофией.
Почки при этом внешне изменены мало, но при тяжелом течении основного патологического процесса они приобретают серовато-желтый цвет, а на разрезе их пирамиды могут принимать желтую окраску.
Исход паренхиматозной жировой дистрофии зависит от степени ее выраженности — пылевидное и мелкокапельное ожирение обратимо при ликвидации вызвавшей его причины, крупнокапельное ожирение может закончиться гибелью клеток.
Врожденные паренхиматозные липидозы
Врожденные паренхиматозные липидозы являются наследственными ферментопатиями, наследуемыми по аутосомно-рецессивному типу, и характеризуются накоплением в клетках липидов, повреждающих структуры клеток и сопровождающихся нередко гибелью самих клеток. Наиболее часто встречаются следующие липидные тезаурисмозы:
- Болезнь Гоше вызвана отсутствием фермента бета-глюкоцереброзидазы. В результате глюкоцереброзиды накапливаются в печени, селезенке, костном мозге, в головном мозге, эндокринных железах и лимфатических узлах, что приводит к гибели клеток этих органов и к прогрессирующему слабоумию, увеличению массы печени, селезенки и истощению (кахексии).
- Болезнь Нимана—Пика развивается при отсутствии фермента сфингомиелиназы, расщепляющей сфингомиелин, входящий в состав многих тканей, но особенно нервной ткани. У больных детей он накапливается в клетках большинства органов и при этом происходит увеличение массы печени и селезенки (гепато- и спленомегалия), отставание в психическом развитии, появляются неврологическая симптоматика, гипотония, истощение. Дети погибают в возрасте 2—3 лет.
ПРИОБРЕТЕННЫЕ Углеводные дистрофии
Гипогликемии — состояния, характеризующиеся снижением содержания глюкозы в крови ниже 65 мг%, или 3,58 ммоль/л. В норме уровень глюкозы крови натощак колеблется в диапазоне 65—110 мг%, или 3,58—6,05 ммоль/л.
Причинами гипоглюкемии являются заболевания печени — xpонические гепатиты, циррозы печени, ее жировая дистрофия, а также длительное голодание.
Результаты заболеваний:
- нарушение транспорта глюкозы из крови в гепатоциты, снижение уровня образования в них гликогенеза и в связи с этим отсутствие депонированного гликогена;
- торможение процесса образования гликогена и транспорта глюкозы из гепатоцитов в кровь.
Последствия гипогликемии
- Гипогликемический синдром — стойкое снижение содержания глюкозы в крови ниже нормы (до 60—50 мг%, или 3,3—2,5 ммоль/л), приводящее к расстройству жизнедеятельности организма.
- Гипогликемическая кома — состояние, характеризующееся:
- — падением концентрации глюкозы в крови ниже 40—30 мг%, или 2.0—1.5 ммоль/л);
- — потерей сознания;
- — опасными для жизни расстройствами функций организма.
Гипергликемии — состояния, характеризующиеся увеличением
содержания глюкозы в крови выше нормы (более 120 мг%, или 6,05 ммоль/л натощак).
Причины гипергликемии:
- патология эндокринной системы, сопровождающаяся избытком гормонов, стимулирующих поступление углеводов в кровь (глюкагона, глюкокортикоидов, катехоламинов, тиреоидных гормонов, соматотропного гормона) либо недостатком инсулина или снижением его эффективности;
- нейро- и психогенные расстройства, например реактивные психозы, стресс-реакции и подобные им состояния, характеризующиеся активацией органов эндокринной системы;
- переедание, прежде всего длительное избыточное потребление кондитерских изделий;
- заболевания печени, при которых гепатоциты теряют cпособность трансформировать глюкозу в гликоген.
Последствия
- Гипергликемический синдром — состояние сопровождающееся значительным увеличением содержания глюкозы в крови выше нормы (до 190—210 мг%, 10,5—11,5 ммоль/л и более), приводящее к расстройствам жизнедеятельности организма.
- Гипергликемическая кома, характеризующаяся потерей сознания, снижением или утратой рефлексов, расстройствами дыхания и кровообращения, нередко заканчивающаяся смертью больного.
Наиболее часто гипергликемия наблюдается при сахарном диабете, развивающемся в результате абсолютной или относительной инсулиновой недостаточности (см. главу 19).
НАСЛЕДСТВЕННЫЕ Углеводные дистрофии (ГЛИКОГЕНОЗЫ)
Гликогенозы — типовая форма патологии углеводного обмена наследственного генеза, характеризующаяся накоплением гликогена в клетках, что обусловливает нарушение жизнедеятельности организма.
причина — наследуемая или врожденная аномалия генов, кодирующих синтез ферментов расщепления (реже — образования) гликогена. Наследуются по аутосомно-рецессивному типу. Выделяют более 10 типов гликогенозов. Среди них наиболее часто встречаются болезни Гирке. Помпе, фетальный муковисцилоз, а также болезни Форбса—Кори, Андерсена, Мак-Ардла.
Болезнь Гирке возникает при отсутствии фермента глюкозо-6-фосфатазы, что приводит к накоплению гликогена в клетках печени и почек, но к отсутствию углеводов в крови. Это сопровождается вторичным гипофизарным ожирением. Большинство детей погибают от ацидотической комы.
Болезнь Помпе связана с отсутствием кислой альфа- 1,4-глюкозидазы в лизосомах, что приводит к накоплению гликогена в сердце, поперечнополосатых и гладких мышцах, в том числе в межреберных, диафрагмальных, в мышцах языка, пищевода, желудка и т.п. Дети погибают в раннем возрасте от сердечной или дыхательной недостаточности.
Остальным муковисцидоз — заболевание, связанное с генотипической ферментопатией. приводящей к нарушению обмена мукоидов, входящих в секрет многих желез.
В результате секрет желез становится вязким и густым, выводится с трудом, что приводит к растяжению желез, превращению их в кисты, особенно в поджелудочной железе, слизистых оболочках желудочно-кишечного тракта и дыхательных путей, слюнных, потовых, слезных железах и др.
При этом в легких часто развиваются ателектазы с развитием пневмонии и бронхоэктазов. Смерть наиболее часто наступает от легочно-сердечной недостаточности., пожалуйста, выделите фрагмент текста и нажмите Ctrl+Enter.
Источник: https://auno.kz/patologiya/258-zhirovye-distrofii-lipidozy.html

